Редкое и очень опасное заболевание - спинальная мышечная атрофия - обычно проявляется у детей первого года жизни. У подростков или взрослых людей подобная патология наблюдается значительно реже.
1
Общее описание болезниСпинальная мышечная атрофия имеет различные формы, которые в свое время были подробно описаны специалистами. При СМА любого вида начинается деформация и/или утрата невронов (нервных клеток) передних рогов спинного мозга. Это нарушает двигательную активность ног, шеи, головы. Больной не может ползать, удерживать голову в вертикальном положении, ходить, глотать пищу. Нормально функционируют только мускулатура верхних конечностей - рук. Чувствительность пораженных участков при этом сохраняется практически полностью. Психиатрической патологии не наблюдаются.
Какие последствия могут быть после проведения спинальной анестезии?
2
Причины развития недугаОсновная причина, вызывающая появление СМА - наследственность. Родители, являющиеся носителями патологической хромосомы, передают ее ребенку. Поэтому чаще всего спинальная мышечная атрофия обнаруживается вскоре после появления малыша на свет.

Патология хромосомы приводит к повреждению (мутации) содержащегося в ней гена. Это нарушает синтез белка. Поэтому двигательная активность снижается, мышечная система атрофируется, постепенно сходят на нет жизненно важные рефлексы (например, глотательный и дыхательный). Дети с врожденной формой СМА в большинстве случаев живут всего 1,5-2 года. Если носителем патологической хромосомы является только 1 из родителей, признаки болезни могут проявиться в отроческом, взрослом или пожилом возрасте, как правило, у мальчиков или мужчин.
Кроме наследственности причиной развития спинальной амиотрофии могут стать такие моменты, как:
Летальный исход в любом случае неизбежен. Однако правильно поставленный диагноз и грамотно составленная схема лечения могут заметно продлить жизнь взрослого пациента.

О чем может говорить боль в груди справа?
3
Классификация СМАОпределение типа болезни у конкретного пациента архиважно. Это определяет схему поддерживающего лечения, виды необходимых процедур, коррекцию режима и т. п. Младенческая спинальная мышечная атрофия или болезнь Верднига -Гоффмана обнаруживается сразу после рождения малыша. Промежуточный тип СМА - болезнь Дубовица появляется у детей через 6-7 месяцев после рождения и до 2-х лет. Спинальная мышечная атрофия ювенильного типа - это болезнь Кугельберга-Веландера. Начинается у детей предподросткового и отроческого возраста. Патология развивается очень быстро.

СМА взрослого типа поражает мужчин среднего и старческого возраста. В этом случае поддерживающая медицина бывает наиболее эффективна.
Основные причины, по которым возникает боль в копчике
4
Болезнь Верднига-ГоффманаБолезнь Верднига- Гофмана можно распознать в период внутриутробного развития младенца. Плод проявляет минимальную активность. Регулярные шевеления в последнем триместре беременности практически отсутствуют. В момент появления на свет ребенок с трудом проходит через родовые пути. Очень часто приходится делать кесарево сечение.
У новорожденного малыша симптомы спинальной мышечной атрофии становятся заметны очень быстро. Ребенок лежит на спине, распластав ноги и руки. Он не может самостоятельно двигать конечностями и поворачивать голову.
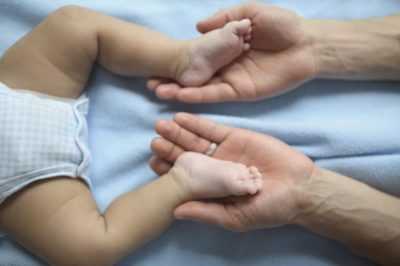
Основной признак младенческой СМА - фасцикуляция. Мышцы, пораженные патологическим процессом, хаотично подергиваются. Может перекосится лицо. У ребенка минимальный сосательный рефлекс. Глотает он с большим трудом. Часто плачет, плохо спит, быстро теряет вес.
5
Как проявляется болезнь Дубовица?Болезнь Дубовица становится заметной после первого полугодия жизни. Атрофия мышц мешает ребенку сидеть и ползать. Такие дети начинают ходить намного позднее. При этом им постоянно требуется помощь.
Нарастание симптоматики в этом случае происходит не так стремительно, как при врожденной СМА. Психическое развитие ребенка проходит нормально. При правильном уходе и постоянном поддерживающем лечении ребенок может прожить более 15 лет.

Ребенок с болезнью Дубовица может самостоятельно себя обслужить, делать простые домашние дела. Такие дети могут учиться в обычной школе, если форма заболевания это позволяет.
При болезни Дубовица сохраняются дыхательный и глотательный рефлексы. Поэтому ребенок может нормально питаться. Такие дети часто страдают от заболеваний верхних дыхательных путей.
Ювенильная форма СМА очень часто проявляется в период полового созревания. В этот момент в организме происходит изменение гормонального фона. Поэтому многие болезни и патологии принимают активную форму.
СМА ювенильного типа развивается медленно. Двигательную активность удается сохранить при помощи специальной гимнастики, физиотерапии и других процедур.
Со временем мускулатура нижних конечностей атрофируется. Больной может передвигаться только в инвалидной коляске. Однако сохраняется определенная степень бытовой самостоятельности.
Согласно медицинской статистике взрослая форма спинальной атрофии мышц наблюдается очень редко по сравнению с другими формами болезни.

Первыми признаками СМА у взрослых людей является:
Больной сохраняет двигательную активность. Он может работать, обслуживать себя, существовать в социуме. Если амиотрофия, начавшаяся у подростка или взрослого мужчины, была обнаружена на ранних стадиях, поддерживающее лечение будет наиболее эффективным.
6
Как диагностировать патологию?Для постановки диагноза проводится анамнестическая беседа с пациентом или опекающим лицом. После общего осмотра больного дополнительно назначается:
После выявления клинической картины определяется схема поддерживающего лечебного курса.
7
Лечение спинальной амиотрофииКардинального средства, позволяющего полностью исцелить больных с диагнозом СМА, не существует. Поэтому при любой форме заболевания основная цель медицинской стратегии - замедление патологических процессов и продление жизни больного. Для этого используются такие препараты, как:

Применение препаратов из указанных групп позволяет нормализовать проведение нервных импульсов, интенсифицировать обменные процессы, ослабляет болевой синдром .
Положительный эффект воздействия лекарственных средств закрепляется при помощи физиотерапии. Чаще всего больным назначается:
Для усиления мышечного тонуса, улучшения циркуляции крови в пораженных областях и предотвращения онемения суставов применяется массаж. Процедура проводится по специальной схеме.
Лечебная гимнастика - обязательное условие сохранения двигательной активности, укрепления костно-мышечной, дыхательной и сердечно-сосудистой системы. Занятия необходимо проводить регулярно с инструктором или самостоятельно. Специальные ортопедические приспособления применяются для поддержания динамики конечностей и нормального функционирования грудной клетки.

Возникающая вследствие болезни дисфункция органов дыхания требует экстренных реанимационных процедур. Пациенту предписывается также искусственная вентиляция легких.
Поддерживающее лечение, специальная гимнастика и другие медицинские процедуры помогают замедлить развитие патологий у взрослых больных. Это позволяет сохранить двигательную активность и рефлексы. Срок жизни заметно увеличивается.
spina-health.com
Содержание:
Спинальная мышечная атрофия – это большая группа врождённых генетических заболеваний, для которых характерно поражение двигательных нейронов передних рогов спинного мозга. То есть происходит поражение особых клеток организма человека, которые отвечают за движение. Впервые болезнь была описана в 1891 году, но чуть позже выяснилось, что существует несколько её форм. Сегодня есть доказательство того, что в одной семье могут рождаться дети с разной степенью поражения двигательных нейронов.
 Спинальные мышечные атрофии, которые во всём мире именуются как SMA – это самая частая генетическая патология, несмотря на то, что этот диагноз ставится довольно редко. Ген болезни был выделен в 1995 году и тогда же он получил название SMN. Из 6 тысяч новорожденных один обязательно рождается с признаками SMA, однако этот показатель различается в разных странах. Большинство детей с диагностированным заболеванием не доживают и до 2 лет.
Спинальные мышечные атрофии, которые во всём мире именуются как SMA – это самая частая генетическая патология, несмотря на то, что этот диагноз ставится довольно редко. Ген болезни был выделен в 1995 году и тогда же он получил название SMN. Из 6 тысяч новорожденных один обязательно рождается с признаками SMA, однако этот показатель различается в разных странах. Большинство детей с диагностированным заболеванием не доживают и до 2 лет.
У этих заболеваний есть некоторые особенности. Одни из них диагностируются практически сразу после рождения крохи, а у других недуг начинает проявляться только в среднем или пожилом возрасте. Такое течение называется «мягким».
По классификации все заболевания, которые относятся к этой группе, можно разделить на несколько групп.
Особенность всех спинальных мышечных атрофий состоит в том, что они все имеют свои собственные проявления.
Это самая тяжёлая форма. Как правило, дети редко доживают до 2 лет. С самого рождения малыши очень слабы, а нередко они не могут сами дышать и даже глотать. При этом пациент должен постоянно находиться на аппарате искусственного дыхания, а питание должно проводиться только через зонд. Умственное и физическое развитие не страдает. Отклонения можно заподозрить ещё при беременности по слабым шевелениям плода.
Дети не могут сидеть из-за атрофии мышц, у них отмечается постоянный тремор пальчиков, отсутствие сухожильных рефлексов, деформация скелета и отсутствие нормального движения в суставе. Часто отмечаются инфекции лёгких и различные дыхательные расстройства.
Для подтверждения диагноза требуется генетическая экспертиза. Вылечить болезнь Верднига-Гоффмана невозможно.
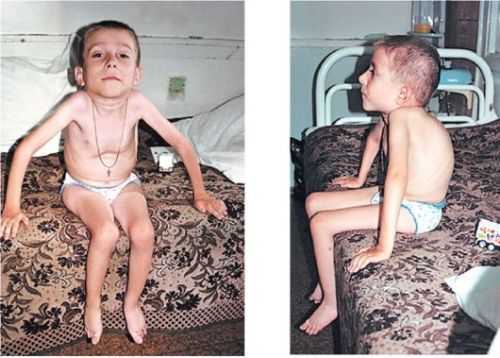
Это заболевание начинает проявлять себя после 6 и до 18 месяцев. При этом недуге также есть слабость в ногах и руках, но она развивается не так быстро. С возрастом руки становятся слабыми, но в них остаётся сила для того, чтобы выполнять какие-то базовые повседневные дела. Ребёнку всю жизнь придётся пользоваться ходунками и лёгкими фиксаторами, которые называются ортезами. При этом он способен управлять электрической инвалидной коляской.
Родители должны особенно пристально следить за дыхательной функцией ребенка и начинать лечить любое заболевание уже в самом начале. Не менее важная проблема – это сколиоз, то есть сильное искривление позвоночника. При этом сколиоз может сильно препятствовать дыханию.
При правильном уходе и родительской заботе дети могут доживать до 16 – 18 лет. Лечения не существует, однако постоянно требуется всех сопутствующих недугов.
Эта разновидность заболевания начинает диагностироваться после того, как ребёнку исполнится 18 месяцев и он начнёт ходить. Многие люди с этим диагнозом могут дожить до 35 – 40 лет, но всё дело в том, что способность передвигаться самостоятельно они теряют уже в юношеском возрасте.
Здесь также большое внимание должно быть приковано к дыханию и искривлению позвоночника. Некоторые пользуются только тросточкой, но многие могут передвигаться только в инвалидной коляске.
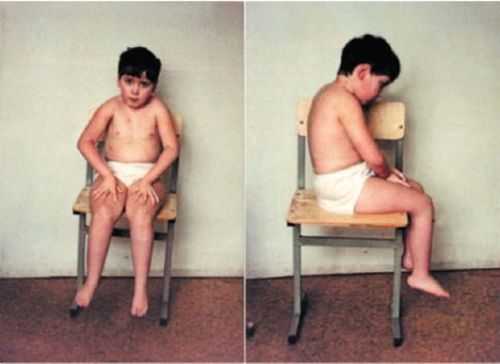
Недуг прогрессирует очень медленно, начиная с поражения нижних конечностей, и постепенно затрагивает верхние. Сухожильные рефлексы угасают постепенно, при этом могут быть отчётливо видны подёргивания в икроножных мышцах или в мышцах плечевого пояса. Человек долго сохраняет способность к самостоятельной жизни.
Введите свой e-mail, чтобы получить бесплатную книгу "7 простых шагов к здоровому позвоночнику"
vashaspina.ru
Запрос СМА перенаправляется сюда. О стиральных машинах см. Стиральная машина
Спина́льная мы́шечная атрофи́я (СМА или SMA) — разнородная группа наследственных заболеваний, протекающих с поражением / потерей моторных нейронов передних рогов спинного мозга.
Для спинальных мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры нижних конечностей, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползание, ходьба, удержание головы, глотание. Мышцы верхних конечностей обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Спинальная мышечная атрофия у детей впервые была описана G. Werdnig в 1891 году. G. Werdnig представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. J. Hoffmann обосновал нозологическую самостоятельность заболевания. В дальнейшем G. Werdnig и J. Hoffmann (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Е. Kugelberg и L. Welander выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной G. Werdnig и J. Hoffmann.
SMA вызвана мутацией в части ДНК, называемой SMN1 ген, который обычно производит белок SMN. Из-за мутации гена, у людей со SMA производится меньшее количество SMN белка, что приводит к потере моторных нейронов.
Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием. Тип 1 SMA проявляется у детей в период от рождения до 6 месяцев.
Пациенты способны сидеть без поддержки или даже стоять с поддержкой и обычно не страдают при приёме пищи. Однако они имеют увеличенный риск осложнений от инфекций дыхательных путей. Тип 2 SMA проявляется у детей в период 7-18 месяцев.
Пациент способен стоять, но испытывает сильную слабость, с тенденцией к инвалидизации (передвижение в коляске). Тип 3 SMA проявляется после 18 месяцев, реже — во взрослом состоянии.
Признаки атрофии обычно начинаются в мышцах рук, ног и языка, затем распространяются к другим областям тела.
Статистика приведена из Patient Registry Report, составитель Connie Garland для SMA Registry, The International Coordinating Committee for clinical trials in SMA:
До последнего времени не было методов специфической терапии СМА.
Так как спинальная мышечная атрофия — нарушение, которое проявляется в синапсах моторных нейронов, состояние может быть улучшено за счёт увеличения уровня SMN — белка. Цель современных исследований — поиск препаратов, увеличивающих уровни SMN. Основные результаты получены пока в исследовательских группах США, Германии, Италии.
Предложено несколько препаратов (вальпроевая кислота, бутират натрия и др.), проводятся их клинические исследования в группах добровольцев. Сведений о результативном применении стволовых клеток пока нет.
Есть сведения, что физиопроцедуры, массаж и др. методы нейро-мышечной стимуляции (напр., дельфинотерапия) положительно сказываются на состоянии больных. В то же время сведений об отрицательных эффектах от физических нагрузок нет.
Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях. Количество вопросов растёт, как снежный ком — только опытный врач может помочь сориентироваться во всех проблемах.
Для объединения больных со спинальной мышечной атрофией (СМА) на Украине инициировано открытие Благотворительного Фонда родителей детей со спинальной атрофией (см. ссылки внизу статьи). Регулярно проводятся телеконференции специалистов и родственников заболевших.
В настоящий момент возможна только пассивная профилактика — предупреждение родителей с риском СМА о возможных последствиях, пренатальный скрининг — диагностика формы СМА для принятия решения о рождении. Диагностика производится в некоторых медико-генетических центрах.
в том числе распространенные заболевания, проявления которых которые могут быть скорректированы путем приема комплекса специальных лекарственных препаратов, диетой, БАД
| Мигрень • Кластерные головные боли • Сосудистая головная боль • Головная боль напряжения |
| Бессонница • Гиперсомния • Апноэ во сне • Нарколепсия • Катаплексия • Синдром Клейне — Левина • Нарушения цикличности сна и бодрствования |
| Дискинезия: Дистония • Хорея • Миоклония • Болезнь Унферрихта — Лундборга • Тремор (Эссенциальный тремор, Интенционный тремор) • Синдром беспокойных ног • Синдром мышечной скованности Заболевания базальных ганглиев: Болезнь Паркинсона • Нейролептический синдром • Пантотенаткиназа-ассоциированная нейродегенерация • Прогрессирующий надъядерный паралич • Стриатонигральная дегенерация • Гемибаллизм |
| Локализованная эпилепсия • Генерализованная эпилепсия • Эпилептический статус • Миоклоническая эпилепсия • Туберозный склероз |
| Болезнь Альцгеймера • Лобно-височная деменция/Лобно-височная лобарная дегенерация • Мультиинфарктная деменция |
| Преходящие нарушения мозгового кровообращения (Гипертензивный церебральный криз, Транзиторная ишемическая атака) • Дисциркуляторная энцефалопатия (Церебральный атеросклероз, Подкорковая атеросклеротическая энцефалопатия, Хроническая гипертоническая энцефалопатия) • Инсульт (Ишемический инсульт, Внутримозговое кровоизлияние, Субарахноидальное кровоизлияние) • Тромбоз синусов твёрдой мозговой оболочки (Тромбоз кавернозного синуса) |
| Абсцесс головного мозга • Менингит • Арахноидит • Энцефалит • Менингоэнцефалит • Энцефалит Расмуссена • Клещевой энцефалит |
| Аутоиммунные заболевания (Рассеянный склероз, Оптикомиелит, Болезнь Шильдера) • Наследственные заболевания (Адренолейкодистрофия, Болезнь Краббе) • Центральный понтинный миелинолиз • Синдром Маркиафавы — Биньями • Синдром Альперса |
| Болезнь Пика • Болезнь Хантингтона • Спинномозговая атаксияСпинальная мышечная атрофия: Синдром Кеннеди • Спинальная мышечная атрофия у детей • Болезнь двигательного нейрона • Синдром Фацио-Лонде • Боковой амиотрофический склероз |
| Синдром Лея |
| Опухоль головного мозга • Туберозный склероз |
| Внутричерепная гипертензия • Отёк мозга • Внутричерепная гипотензия |
| Черепно-мозговая травма (Сотрясение мозга, Ушиб головного мозга, Диффузное аксональное повреждение головного мозга) |
| Спинномозговая грыжа • Синдром Рея • Печёночная кома • Токсическая энцефалопатия • Гематомиелия |
| Менингит • Арахноидит • Менингоэнцефалит • Миелит • Полиомиелит • Демиелинизирующие заболевания • Тропический спастический парапарез |
| Сирингомиелия • Сирингобульбия • Синдром Морвана • Сосудитая миелопатия • Спинальный инсульт • Сдавление спинного мозга • Энцефаломиелит |
dic.academic.ru
ЧТО ТАКОЕ - СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ? Спинальная мышечная атрофия (СМА) — генетическое заболевание, поражающее область нервной системы, отвечающей за контроль движений произвольных (скелетных) мышц
Большинство нервных клеток, контролирующих мышцы, находятся в спинном мозге, отсюда — слово «спинальная» в названии болезни. Слово «мышечная» говорит о том, что страдают мышцы, которые не получают сигналов от этих нервных клеток. Ну и «атрофия» - медицинский термин для истощения или «усыхания», которое происходит с мышцами, когда они неактивны
При СМА происходит утрата нервных клеток в спинном мозге, именуемых мотонейронами, поэтому она классифицируется как болезнь мотонейронов
Возраст начала и тяжесть течения СМА варьируется у разных людей в широком диапазоне
Что является причиной СМА?
В большинство случаев СМА обусловлена дефицитом протеина мотонейронов, именуемым SMN (протеин выживаемости мотонейронов)
Этот протеин, как следует из его названия, необходим для нормального функционирования мотонейронов. Недавние исследования наводят также на мысль о том, что отсутствие SMN может также непосредственно влиять на мышечные клетки
Существуют также формы СМА, не связанные с протеином SMN
КАКИЕ ЕСТЬ ФОРМЫ СМА?
SMN-связанная СМА
SMN – связанная СМА обычно подразделяется на 3 категории. Тип 1 — наиболее тяжелый, с самым ранним началом, а тип 3 — наименее тяжелый, с наиболее поздним возрастом начала. Некоторые специалисты выделяют еще тип 4 для обозначения умеренной или мягкой СМА с дебютом во взрослом возрасте
Все эти типы связаны с генетическими поломками (мутациями) на хромосоме 5, что влияет на количество синтезируемого SMN – протеина. Более высокие уровни протеина снижают тяжесть СМА. Более подробно о том, как эти мутации приводят к развитию СМА, описано в разделе «Это семейное?»
Не связанная с SMN СМА
Существуют также формы СМА, не связанные с SMN и не являющиеся результатом мутаций на хромосоме 5. Подробнее об этом — в разделе «Что происходит с людьми с различными формами СМА?»
Спино-бульбарная мышечная атрофия (СБМА)
Этот тип СМА, именуемый также болезнью Кеннеди, является результатом мутаций гена на Х-хромосоме, и значительно отличается от SMN-связанных типов СМА. Более подробно — в соответствующем разделе.
ЧТО ПРОИСХОДИТ С ЛЮДЬМИ, ИМЕЮЩИМИ SMN-СВЯЗАННЫЕ ФОРМЫ СМА?
Тяжесть SMN-связанных СМА зависит от того, насколько рано проявляются симптомы, что в свою очередь взаимосвязано с количеством SMN протеина в мотонейронах. Более позднее появление симптомов и более высокие уровни SMN протеина предполагают, что течение заболевание будет более мягким
Однако в настоящее время большинство специалистов сомневаются в столь строгой зависимости и предпочитают не делать однозначных прогнозов по тяжести течения и продолжительности жизни, основываясь только на возрасте дебюта. Последние исследования говорят в поддержку такой гибкости
СМА тип 1 (болезнь Верднига-Гоффмана)
Если дети с СМА очень слабы в первые месяцы жизни и испытывают трудности с дыханием, сосанием и глотанием, по всей видимости трудно рассчитывать для них на хороший прогноз. Ранее считалось, что такие дети не выживают более двух лет. И в настоящее время в большинстве случаев такой прогноз оказывается справедливым
Тем не менее с применением технологий взамен естественных функций дыхания и питания такие дети могут выживать в течение нескольких лет. Механическая вентиляция легких (сейчас такие аппараты стали портативными, в отличие от применявшихся в предыдущие годы «железных легких» и тяжелых механизмов) и питание через зонды непосредственно в желудок, а не в глотку, могут продлить жизнь
Умственное и эмоциональное развитие, а также чувствительность при СМА совершенно нормальны
СМА тип 2 (промежуточная СМА)
Диагноз СМА тип 2 позволяет родителям и детям планировать будущее, несмотря на сокращенную продолжительность жизни. Этот тип СМА дебютирует в детском возрасте, но как правило позже младенчества. В некоторых источниках к типу 2 относят заболевание с возрастом дебюта в промежутке между 6 и 12 месяцами. В других источниках говорится, что заболевание классифицируется как тип 2, если ребенок способен сидеть без поддержки, если его посадить
При этом типе СМА мышцы, расположенные ближе к центру туловища (т.н. проксимальные мышцы) обычно поражены в большей степени, или по крайней мере поражаются раньше, чем мышцы, более удаленные от центра (дистальные). Например, мышцы бедер слабее, чем мышцы голеней и стоп.
Кроме того, ноги обычно ослабевают раньше, чем руки. Руки в итоге ослабевают, но все же остаются более сильными, и, даже если ослабевают, обычно сохраняют силу, достаточную для печати на клавиатуре компьютера и для выполнения других базовых функций современной жизни.
Дети с СМА типа 2 получают большую выгоду от физиотерапии и применения различных вспомогательных приспособлений всех видов. Приспособления для стояния и хождения, такие как легкие фиксаторы (ортезы) и ходунки, дают бОльшую мобильность, чем это было возможно в прошлом.
Многие дети способны управлять электроколясками или другими средствами передвижения довольно рано, уже в возрасте 3 лет или около того в зависимости от степени развития. Многие специалисты отмечают, что дети с СМА необычайно умны и сообразительны, а немногочисленные исследования подтверждают эти наблюдения
Наибольшую опасность при этом типе СМА представляет слабость дыхательных мышц. На протяжении всей жизни необходимо пристальное внимание к дыхательной функции и немедленное реагирование на инфекции. Ваш врач может помочь вам с особыми методами поддержания здоровья дыхательной системы, включая очистку дыхательных путей от выделений и возможную вентиляцию легких. Подробнее об этом см. «Слабость дыхательной мускулатуры»
Другая серьезна проблема при этом типе СМА — искривление позвоночника, обычно — в поперечном направлении, именуемое сколиозом. Сколиоз развивается из-за слабости мышц, поддерживающих позвоночник, который является гибким столбом. Сколиоз причиняет большие неудобства, препятствуя сохранению нормального положения тела и мобильности, и серьезно портя вид тела ребенка (или взрослого) Некоторые исследования показывают также, что тяжелое искривление позвоночника препятствует и дыханию
У многих детей с СМА сколиотическое искривление проявляется уже в начале жизни, и как правило корректируется с помощью фиксаторов до тех пор, пока не наступит подходящее время для хирургического вмешательства. Хирурги обычно склонны подождать, пока рост полностью (или почти) не прекратится, прежде чем хирургическим путем выправить и закрепить позвоночник. Они также наблюдают за дыхательной функцией ребенка и за тем, как быстро идет прогресс искривления
В настоящее время продолжительность жизни при СМА с дебютом в детстве варьируется. Вполне вероятно выживание до юношеского возраста и даже дольше
СМА тип 3 (болезнь Кугельберга-Веландер или умеренная СМА)
Некоторые источники описывают СМА тип 3 как СМА, начинающуюся в возрасте старше 18 месяцев, в то время как другие предпочитают говорить о ней как о СМА, начинающуюся после того, как ребенок начал ходить, или по крайней мере сделал самостоятельно пять шагов
Многие люди с этим типом СМА способны ходить до 30-40 лет, хотя некоторые теряют способность к хождению уже в юности
Важным остается пристальное внимание к дыханию и потенциальному искривлению позвоночника. При этом типе СМА обычно важны вспомогательные приспособления, такие как электроколяски, устройства для пользования компьютером и т. п. Некоторые люди обходятся только тростью и, иногда, складными стульчиками, а колясками пользуются только для передвижения на бОльшие расстояния, например в аэропорту или торговом центре
Люди с этим типом СМА доживают до зрелого возраста и достигают успехов в научной и трудовой деятельности
СМА тип 4 (СМА с дебютом во взрослом возрасте)
Этот тип СМА остается мягким на всем своем протяжении. По определению, заболевание при этом типе дебютирует в зрелом возрасте. Некоторые специалисты объединяют вместе типы 3 и 4, или типы 2 и 3
ЧТО ПРОИСХОДИТ С ЛЮДЬМИ С ДРУГИМИ ФОРМАМИ СМА?
Другие типы СМА, не связанные с дефицитом протеина SMN, значительно отличаются по тяжести и наиболее пораженным мышцам. При некоторых формах СМА, подобных SMN-связанным типам, наиболее поражены проксимальные мышцы, в то время как при других — наоборот дистальные, т. е. те, которые наиболее удалены от центра тела, по крайней мере в начале. На ранней стадии как правило поражаются кисти рук и стопы
Иногда у взрослых с заболеванием неясного происхождения, поражающим только мотонейроны спинного мозга и нижней части головного мозга, состояние относят к прогрессирующей мышечной атрофии. Это состояние иногда прогрессирует с вовлечением мотонейронов верхней части головного мозга, и тогда это состояние определяется как боковой амиотрофический склероз (БАС) Трансформация этого состояния в БАС происходит не всегда
Особенности спино-бульбарной мышечной атрофии (СБМА) рассмотрены ниже
Хотя все известные формы СМА — генетические, они являются результатом дефектов в различных генах и имеют разные типы наследования и прогнозы при планировании семьи
Если вам или вашему ребенку поставлен диагноз СМА, но не SMN-связанный тип, вам необходимо поговорить с вашим врачом, а возможно — и с генетиком-консультантом, чтобы больше узнать о генетике и прогнозах при этом определенном типе СМА
КАК СМА ЛЕЧИТСЯ?
Наибольшие потенциальные проблемы при СМА, особенно при формах, связанных с 5 хромосомой, в приблизительном порядке по серьезности, это:
слабость дыхательных мышцслабость глотательных мышцслабость мышц спины с прогрессирующим искривлением позвоночникааномальная реакция на препараты-миорелаксанты
Эти проблемы можно, и необходимо, лечить и предупреждать
Слабость дыхательных мышц
При наиболее тяжелых формах SMN-связанных СМА, и при некоторых других формах, слабость дыхательных мышц является серьезной проблемой. Это основная причина гибели детей с СМА типа 1 и 2. Когда дыхательные мышцы ослаблены, воздух не может хорошо циркулировать в легких, что негативно сказывается на общем здоровье. Симптомами слабости дыхательной мускулатуры являются головная боль, трудности со сном в ночное время, чрезмерная сонливость в дневное время, плохая концентрация, инфекции грудной клетки и, в конечном итоге, сердечная и дыхательная недостаточность
При СМА тип 1 очень ослаблены межреберные мышцы, в то время как мышцы диафрагмы остаются довольно сильными. В этом случае дети вынуждены дышать больше с помощью движений живота, чем грудной клетки, что придает их телу грушевидную форму
Родители новорожденных с СМА 1 типа могут столкнуться с вопросом, как продлить жизнь своего ребенка дольше, чем предполагаемые при этом типе два года. В последние годы доступность портативных и эффективных устройств для вентиляции легких дает родителям больше возможностей для этого. Некоторые такие дети к удивлению своих семей и докторов живут много лет, иногда — до подросткового возраста
Различные типы устройств вентиляции могут помочь взрослым и детям с менее тяжелым поражением. Многие специалисты рекомендуют начинать с неинвазивной вентиляции, которая как правило означает, что воздух (обычно комнатный воздух, не обогащенный кислородом) подается под давлением через маску или мундштук
Системы такого типа поставляются во многих формах и могут использоваться при необходимости по многу часов днем и/или ночью. Они легко могут быть сняты для еды, питья, разговора и, когда это возможно, для дыхания без их помощи. Некоторые пациенты предпочитают использовать вентиляционные системы с отрицательным давлением (разрежением), которые создают периодический вакуум вокруг грудной клетки или туловища, что помогает легким расширятся и сжиматься. Эти устройства работают по тому же принципу, что и применявшиеся несколько десятилетий назад т. н. «железные легкие», но при этом существенно менее громоздки
Для детей и взрослых с тяжелым поражением часто рекомендуется вентиляция через трахеостому — сделанное хирургическим путем отверстие в трахее, или дыхательном горле. В этом случае воздух под давление подается по трубке в трахеостоме. Обычно люди могут есть, пить и разговаривать с трубкой в трахеостоме, хотя этот процесс и требует некоторой адаптации. Существует некоторое беспокойство, что если ребенку делают трахеостому до того, как он начал говорить, это может вызвать сложности с его обучением речи
Другими важными аспектами решения дыхательных проблем при СМА является очистка дыхательных путей, для которой также иногда применяются механические устройства, и предотвращение инфекций столько, сколько это возможно
Слабость глотательных мышц
Слабость мышц рта и глотки вызывает проблемы с глотанием, особенно при наиболее тяжелых формах СМА. Дети со СМА тип 1 обычно испытывают трудности с глотанием и сосанием, что впоследствии приводит к их гибели. Слабость сосания приводит к обезвоживанию и недостаточному питанию, а трудности с глотанием — к непроходимости дыхательных путей вследствие вдыхания пищи или жидкости (это называется аспирацией) и респираторным инфекциям.
В наши дни питание детей с трудностями глотания можно осуществлять альтернативными методами, такими, как гастростомическая трубка (g-трубка). Конструкция современных систем позволяет отсоединять трубку от «кнопки» на брюшине, когда она не используется. Жидкая пища, широко доступная в магазинах, подается в трубку с помощью шприца или питательного насоса. Некоторые люди просто измельчают обычную пищу
Некоторые пользователи g-трубки также едят и пьют обычным способом. Если основная проблема — слабость жевательных мышц, то процесс еды труден и продолжителен по времени, но в то же время приятно поесть для удовольствия и дополнительного питания, получая основные калории через g-трубку. Однако если основной причиной применения питательной трубки является постоянное вдыхание еды и пищи, вероятно не стоит пробовать есть и пить обычным способом
При СБМА слабость глотательных и жевательных мышц вызывает риск удушья. Необходимо проконсультироваться у специалиста для определения наиболее безопасных способов дыхания и консистенции пищи. В случае серьезной слабости необходимо рассмотреть применение питательной трубки
Слабость мышц спины
Слабость мышц спины, в норме осуществляющих поддержку гибкого и высокого позвоночника — основная проблема при СМА с дебютом в детстве. Если не уделять этому внимания, у ребенка может развиться сколиоз (искривление позвоночника в поперечном направлении) или кифоз (искривление в продольном направлении), или и то и другое. Иногда это может закончиться «кренделеобразным» искривлением, что делает невозможным комфортное сидение и лежание
Многие специалисты уверены, что тяжелое искривление позвоночника негативно влияет и на дыхательную функцию, поскольку искривленный позвоночник часто давит на легкие. Однако при тяжелых формах СМА трудно предугадать, насколько ухудшится дыхательная функция, даже если искривления не будет, поэтому его вклад в это неопределен
Закрепление спины с помощью фиксаторов или корсетов для поддержки ребенка в определенном положении часто применяется в попытке направлять позвоночник в процессе его роста. Такие приспособления не решают проблему, хотя могут замедлить прогрессирование искривления, что желательно
Окончательным итогом искривления позвоночника является хирургическое вмешательствооо по его закреплению (т. н. спондилодез), которое может быть произведено, если респираторный статус ребенка позволяет ему перенести такое вмешательство. Как правило доктора предпочитают подождать до окончания роста позвоночника, чтобы можно было использовать простейшую хирургическую технику. Однако не всегда возможно дождаться окончания роста, поскольку респираторный статус может ухудшится в любой момент. Поэтому выбор момента для такой операции — непростая задача
Несколько слов об анестезии
Детям и взрослым, которым необходимо хирургическое вмешательство (например, для коррекции сколиоза), нужно соблюдать специальные меры предосторожности. Хирургическая бригада, особенно анестезиолог, должна хорошо понимать, что такое СМА
Иногда, особенно на ранних стадиях СМА, в мышечных клетках, не получающих сигналов от нервов, развиваются определенные аномалии, поскольку они пытаются «дотянуться» до нервов. Эти аномалии могут вывать опасную реакцию на миорелаксанты, часто применяемые при операциях. Если знать о возможности таких проблем, их можно избежать применением различных препаратов
Поговорим о диете
Многие люди задумываются, не сможет ли какое то специальное питание их детей или их самих повлиять на течение болезни. Однако, пока нет доказательств того, что какая то определенная диета полезна при СМА
Многие родители полагают, что высоко-белковая диета, или какие либо виды специальных пищевых добавок смогут помочь мышцам их детей стать сильнее. Однако хотя не вызывает сомнений, что детям со СМА необходимо хорошее питание, не существует подтверждений, что необходим какой-то определенный тип питания, более того — что-то может быть и вредным
Например, специальные формулы, состоящие из компонентов протеинов, именуемых аминокислотами - так называемые «элементные диеты» - могут в действительности привести к проблемам у детей со СМА, имеющих мало мышечной ткани. Некоторые эксперты отмечают, что эти аминокислоты могут подняться до слишком высоких уровней в крови, если объема мышечной ткани недостаточно для их правильной переработки
Некоторым детям может быть более полезно частое питание маленькими порциями, чем трехразовое питание с большими порциями
Некоторые дети и многие взрослые с СМА имеют избыточный вес, вероятно из-за того, что получают слишком много калорий для своего уровня физической активности. По возможности следует под руководством врача и диетолога держать вес под контролем, что важно для здоровья, внешнего вида, а также для спин ухаживающих, которые ежедневно помогают с подъемом и перемещением
Некоторые специалисты рекомендуют употребление безрецептурных пищевых добавок, таких как креатин или коэнзим Q10. Применение креатина при СМА исследуется
Какие упражнения лучше всего?
Большинство специалистов говорят людям со СМА и родителям детей со СМА, что комфортная физическая активность без впадания в крайности — хорошая идея для физического и психологического здоровья и самочувствия
Важно предохранять суставы от неподвижности и повреждений, сохраняя объем движений (эластичность суставов), поддерживая циркуляцию и, что особенно важно для детей, поддерживая мобильность на достаточном уровне для исследования окружающего пространства
Особенно полезными могут быть занятия в бассейне с теплой водой (30-32 град). Люди с СМА не должны плавать самостоятельно, и должны быть соблюдены необходимые меры безопасности
Некоторые специалисты поднимают вопрос о том, стоит ли уделять слишком много внимания постепенному уменьшению количества мотонейронов, которым приходится выполнять работу, которая в норме выполняется гораздо бОльшим количеством этих клеток. Необходимы исследования, чтобы определить, нужно ли в действительности учитывать эту проблему при разработке плана упражнений. Некоторые эксперты полагают, что перегрузка невозможна, в то время как другие считают, что упражнения до изнеможения могу «сжечь» оставшиеся мотонейроны раньше времени. Поэтому представляется целесообразным заниматься физическими упражнениями с осторожностью и прекращать их до наступления изнеможения
Физио- и трудотерапевтические программы могут помочь как детям, так и взрослым научиться наилучшему использованию имеющихся у них мышечных функций, и найти наиболее эффективные способы выполнения повседневных дел
Существующие сегодня различные вспомогательны приспособления могут помочь даже самым маленьким детям в познании окружающего мира. Вертикализаторы, ходунки, различные типы электрических и ручных колясок, ортезы — все это может помочь в стоянии и перемещении.
Некоторые семьи даже сами разрабатывают и делают свои собственные приспособления со специальными возможностями, например — с регулируемой высотой, позволяющей детям как двигаться на уровне пола, так и сидеть на столом
Полезными как для взрослых, так и для детей со связанной с СМА слабостью могут быть и специальные приспособления для письма, рисования, использования компьютера или телефона, и электронные системы контроля окружающей обстановки (например — температуры, освещенности, управление телевизором и т.п.)
КАК СМА ДИАГНОСТИРУЕТСЯ?
Первый шаг в диагностике нервно-мышечных заболеваний — осмотр в кабинете врача и изучение семейной истории для того, чтобы отличить СМА от схожих заболеваний (таких как мышечная дистрофия)
Доктор может назначить исследование крови на энзим, именуемый креатинкиназой (КФК) Этот энзим просачивается из мышц про их повреждении. Это неспецифический тест, поскольку повышенные уровни КФК бывают при многих нервно мышечных заболеваниях, тем не менее он часто используется. Повышенный уровень КФК сам по себе неопасен, это всего лишь индикатор повреждения мышц
При подозрении на СМА врач вероятно порекомендует генетическое тестирование, поскольку это наиболее безболезненный и точный способ диагностики SMN-связанных типов СМА, а также СБМА
Для генетического тестирования требуется только образец крови. Однако это важно для всей семьи (См. «Это семейное?»)
Если генетические тесты для SMN-связанных форм и СБМА легко доступны, то тесты для более редких форм СМА по большей части доступны только в рамках исследований. Однако такие тесты быстро совершенствуются и распространяются по мере улучшения знаний и технологий. В ближайшем будущем вероятно выскодетализированные генетические тесты SMN-связанных СМА можно будет использовать для гораздо более точного прогнозирования течения болезни, чем это возможно сейчас
sma-family.livejournal.com
НАШИ ЧИТАТЕЛИ РЕКОМЕНДУЮТ!
Для лечения суставов наши читатели успешно используют Артрейд. Видя, такую популярность этого средства мы решили предложить его и вашему вниманию.Подробнее здесь…
Спинальная мышечная атрофия (амиотрофия) – это группа генетических заболеваний, при которых развивается прогрессирующая атрофия мышц в результате поражения нейронов передних рогов спинного мозга. Болезнь чаще развивается в первые годы после рождения и имеет неблагоприятный исход. Возникновение патологического процесса в зрелом возрасте характеризуется медленным нарастанием симптомов и более благоприятным течением. До сих пор не найдено эффективное лечение спинальной атрофии мышц. Однако учеными многих стран мира ведутся медицинские разработки лекарственного препарата, который сможет стать прорывом в терапии тяжелой неврологической болезни.
Амиотрофия развивается при мутации гена в 5 хромосоме, который получил название SMN (survival motor neuron). Он передается по аутосомно-рецессивному типу и вызывает развитие болезни только при получении измененного генетического материала от обоих родителей, что бывает довольно редко.
Носительство гена не влияет на состояние здоровья. Научными исследованиями доказано, что каждый 40-й человек имеет мутацию гена мышечной атрофии.
Ген SMN влияет на выработку специфического белка, который необходим для жизнедеятельности мотонейронов, расположенных в передних рогах спинного мозга. В результате нервные клетки не выполняют основные функции – проведение нервных импульсов из центральных отделов на периферию к мышечным волокнам. Изменение иннервации мышц приводит к нарушению кровотока, обменных процессов и сократимости, что со временем вызывает их ослабление, уменьшение в объеме, неспособность поддерживать нормальную двигательную активность.
Гибель двигательных нейронов происходит в передних отделах спинного мозга, при этом имеет симметричную локализацию. Этот характерный признак рассматривается в пользу диагноза спинальной мышечной атрофии. В некоторых случаях не удается подтвердить наследственный характер заболевания. Приобретенные формы болезни могут возникать в результате травм спинного мозга, инфекционного поражения центральной нервной системы сифилисом, эндокринных нарушений при сахарном диабете, авитаминозе.
Амиотрофия имеет несколько форм, которые отличаются возрастом появления первых симптомов, тяжестью течения патологического процесса и продолжительностью жизни больных. Заболевание характеризуется развитием инвалидности, которая сопровождается нарушением двигательной активности и неспособностью к самообслуживанию в быту. В тяжелых клинических случаях больные требуют постоянного врачебного наблюдения и посторонней помощи в повседневной жизни.
Передвижение возможно с помощью инвалидных кресел, ходунков, костылей, трости. Летальный исход наступает вследствие застойных осложнений со стороны дыхательной и сердечно-сосудистой системы, которые проявляются тяжелыми пневмониями и сердечной недостаточностью. В патологический процесс не вовлекаются чувствительные нервные волокна, поэтому сохраняются все виды чувствительности. Интеллект и ментальные функции не страдают и при обучении ребенка хорошо развиты.
Болезнь Верднига-Гоффмана была открыта в конце 18-го века известными врачами-невропатологами, в честь которых она и была названа. Они описали самый тяжелый 1 тип заболевания с неблагоприятным прогнозом и значительными нарушениями в двигательной активности конечностей. Во времена открытия болезни и по сей день прогноз для жизни больных остается неутешительным, часто пациенты умирают в раннем детском возрасте.
Патологический процесс в спинном мозге может развиваться во время внутриутробного периода. В таких случаях обнаруживают слабую активность плода и редкое шевеление во второй половине беременности. После рождения симптомы заболевания нарастают на протяжении первых 6-ти месяцев жизни ребенка. Отмечают вялость двигательной активности – новорожденный обычно лежит в распластанной позе на спине (поза «лягушки»), не переворачивается, редко сгибает и разгибает ножки. Характерный симптом заболевания – хаотичное подергивание пораженных мышц, которое носит название фасцикуляция.
В результате повреждения мотонейронов и нарушения поступления нервных импульсов из головного мозга мышцы не развиваются, уменьшаются в объеме. Атрофия мышечных волокон может скрываться под хорошо развитым подкожно-жировым слоем в начале развития болезни. При обследовании не определяют сухожильные рефлексы нижних, реже верхних конечностей. Возникают проблемы с дыханием и глотанием вследствие нарушения иннервации глотки, межреберных и брюшных мышц, диафрагмы. Это вызывает появление застойной и аспирационной пневмонии, что при несвоевременной диагностике приводит к летальному исходу.
Болезнь Верднига-Гоффмана характеризуется деформацией скелета в силу неспособности ослабленных мышц удерживать кости в анатомически правильном положении. При попытках ребенка сидеть формируется искривление позвоночника, развивается сколиоз и патологический кифоз. Атрофия межреберных мышц приводит к уплощению грудной клетки, что в свою очередь усугубляет работу сердца и органов дыхания. В начале болезни поражается мускулатура нижних конечностей, вследствие этого ребенок не может становиться на ноги, не способен ходить, на протяжении всей жизни прикован к инвалидному креслу. Атрофия мышц головы и шеи затрудняет движения головой, а снижение силы верхних конечностей не дает развиваться навыкам самообслуживания в быту.
Младенческая форма или 2 тип спинальной мышечной атрофии формируется у детей в возрасте 7-18 месяцев и диагностируется при попытках ребенка ползать, сидеть, ходить самостоятельно. С самого рождения отмечают вялую двигательную активность новорожденных, задержку в физическом развитии, снижение сухожильных рефлексов. Младенческая форма протекает легче 1 типа заболевания, характеризуется медленным прогрессированием атрофии мышц, продолжительность жизни больных обычно достигает 16-18 лет.
Дети способны самостоятельно сидеть, ползать, вставать на ноги, но не могут ходить без посторонней помощи. В быту сохраняются навыки самообслуживания, что также дает возможность приобщить детей к самостоятельному обучению. Дыхательная деятельность не затруднена, глотательные рефлексы сохранены. Однако в силу низкой двигательной активности пациенты уязвимы к респираторным заболеваниям и воспалению легких, что значительно ухудшает здоровье и может привести к тяжелым осложнениям.
Болезнь Куленберга-Веландера или ювенильная форма возникает после достижения двухлетнего возраста, часто развивается в период полового созревания. Характеризуется медленным прогрессирующим течением. Такие больные в начале развития патологии способны самостоятельно ходить, подниматься по лестнице, садиться на стул или кровать и вставать.
Однако со временем двигаться становится труднее, для выполнения элементарных движений приходится прибегать к значительным усилиям. В результате гипертрофируются (утолщаются) мышцы бедра и ягодиц. В первую очередь страдают мышцы ног, затем в патологический процесс вовлекаются мышечные группы спины, шеи, верхних конечностей. Способность к передвижению и самообслуживанию сохраняется довольно длительное время, но неизбежным исходом заболевания является инвалидность – больные вынуждены пользоваться инвалидной коляской.
Взрослая форма болезни встречается гораздо реже вышеперечисленных типов заболевания и развивается в возрасте старше 30-40 лет. Она характеризуется медленным прогрессированием и благоприятным исходом. В патологический процесс вовлекаются преимущественно мышцы головы и шеи, что не влияет на потерю трудоспособности и не приводит к нарушению двигательной активности. Характерным клиническим признаком считается подергивание языка, слабая мимика и ограничение подвижности головы. Чем позже появляются первые симптомы амиотрофии, тем благоприятнее прогноз для жизни.
Диагноз заболевания устанавливают по характерным клиническим признакам и результатам неврологического обследования. Подтверждают амиотрофию путем биопсии пораженных мышц, где обнаруживают патологические изменения. Проводят компьютерную и магнитно-резонансную томографию (КТ, МРТ) для обнаружения нарушений в области передних отделов спинного мозга. Назначают нейромиографию с целью выявления замедления или отсутствия процессов передачи нервных импульсов.
Следует понимать, что современная медицина не может вылечить заболевание. Терапия направлена на поддержание жизненных функций больного и предупреждение осложнений. Консервативное лечение включает:
В терминальных стадиях развития болезни могут понадобиться реанимационные мероприятия и перевод на искусственную вентиляцию легких вследствие необратимых изменений мышц, участвующих в процессе дыхания.
Спинальная мышечная атрофия относится к тяжелому неврологическому заболеванию, связанному с поражением двигательных путей передних рогов спинного мозга. Разработки инновационного препарата, который сможет восполнить недостающий нейронный белок, дает надежду больным на выздоровление.
osteo.lechenie-sustavy.ru
НАШИ ЧИТАТЕЛИ РЕКОМЕНДУЮТ!
Для лечения суставов наши читатели успешно используют Артрейд. Видя, такую популярность этого средства мы решили предложить его и вашему вниманию.Подробнее здесь…
Содержание:
Спинальная мышечная атрофия – это большая группа врождённых генетических заболеваний, для которых характерно поражение двигательных нейронов передних рогов спинного мозга. То есть происходит поражение особых клеток организма человека, которые отвечают за движение. Впервые болезнь была описана в 1891 году, но чуть позже выяснилось, что существует несколько её форм. Сегодня есть доказательство того, что в одной семье могут рождаться дети с разной степенью поражения двигательных нейронов.
 Спинальные мышечные атрофии, которые во всём мире именуются как SMA – это самая частая генетическая патология, несмотря на то, что этот диагноз ставится довольно редко. Ген болезни был выделен в 1995 году и тогда же он получил название SMN. Из 6 тысяч новорожденных один обязательно рождается с признаками SMA, однако этот показатель различается в разных странах. Большинство детей с диагностированным заболеванием не доживают и до 2 лет.
Спинальные мышечные атрофии, которые во всём мире именуются как SMA – это самая частая генетическая патология, несмотря на то, что этот диагноз ставится довольно редко. Ген болезни был выделен в 1995 году и тогда же он получил название SMN. Из 6 тысяч новорожденных один обязательно рождается с признаками SMA, однако этот показатель различается в разных странах. Большинство детей с диагностированным заболеванием не доживают и до 2 лет.
У этих заболеваний есть некоторые особенности. Одни из них диагностируются практически сразу после рождения крохи, а у других недуг начинает проявляться только в среднем или пожилом возрасте. Такое течение называется «мягким».
По классификации все заболевания, которые относятся к этой группе, можно разделить на несколько групп.
Особенность всех спинальных мышечных атрофий состоит в том, что они все имеют свои собственные проявления.
Это самая тяжёлая форма. Как правило, дети редко доживают до 2 лет. С самого рождения малыши очень слабы, а нередко они не могут сами дышать и даже глотать. При этом пациент должен постоянно находиться на аппарате искусственного дыхания, а питание должно проводиться только через зонд. Умственное и физическое развитие не страдает. Отклонения можно заподозрить ещё при беременности по слабым шевелениям плода.
Дети не могут сидеть из-за атрофии мышц, у них отмечается постоянный тремор пальчиков, отсутствие сухожильных рефлексов, деформация скелета и отсутствие нормального движения в суставе. Часто отмечаются инфекции лёгких и различные дыхательные расстройства.
Для подтверждения диагноза требуется генетическая экспертиза. Вылечить болезнь Верднига-Гоффмана невозможно.
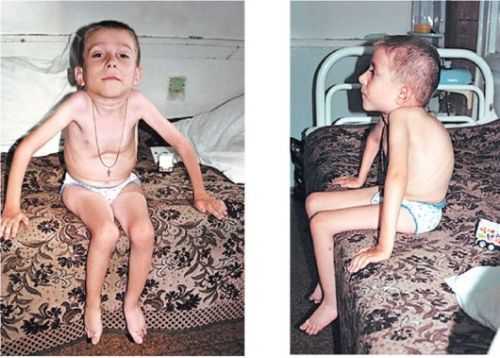
Это заболевание начинает проявлять себя после 6 и до 18 месяцев. При этом недуге также есть слабость в ногах и руках, но она развивается не так быстро. С возрастом руки становятся слабыми, но в них остаётся сила для того, чтобы выполнять какие-то базовые повседневные дела. Ребёнку всю жизнь придётся пользоваться ходунками и лёгкими фиксаторами, которые называются ортезами. При этом он способен управлять электрической инвалидной коляской.
Родители должны особенно пристально следить за дыхательной функцией ребенка и начинать лечить любое заболевание уже в самом начале. Не менее важная проблема – это сколиоз, то есть сильное искривление позвоночника. При этом сколиоз может сильно препятствовать дыханию.
При правильном уходе и родительской заботе дети могут доживать до 16 – 18 лет. Лечения не существует, однако постоянно требуется всех сопутствующих недугов.
Эта разновидность заболевания начинает диагностироваться после того, как ребёнку исполнится 18 месяцев и он начнёт ходить. Многие люди с этим диагнозом могут дожить до 35 – 40 лет, но всё дело в том, что способность передвигаться самостоятельно они теряют уже в юношеском возрасте.
Здесь также большое внимание должно быть приковано к дыханию и искривлению позвоночника. Некоторые пользуются только тросточкой, но многие могут передвигаться только в инвалидной коляске.

Недуг прогрессирует очень медленно, начиная с поражения нижних конечностей, и постепенно затрагивает верхние. Сухожильные рефлексы угасают постепенно, при этом могут быть отчётливо видны подёргивания в икроножных мышцах или в мышцах плечевого пояса. Человек долго сохраняет способность к самостоятельной жизни.
pozvon.lechenie-sustavy.ru
Спина́льная мы́шечная атрофи́я (СМА или SMA) — разнородная группа наследственных заболеваний, протекающих с поражением / потерей моторных нейронов передних рогов спинного мозга.
Для спинальных мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры нижних конечностей, а также головы и шеи. У больных отмечаются нарушения произвольных движений - ползание, ходьба, удержание головы, глотание. Мышцы верхних конечностей обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Спинальная мышечная атрофия у детей впервые была описана G. Werdnig в 1891 году. G. Werdnig представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. J. Hoffmann обосновал нозологическую самостоятельность заболевания. В дальнейшем G. Werdnig и J. Hoffmann (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Е. Kugelberg и L. Welander выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной G. Werdnig и J. Hoffmann.
SMA вызвана мутацией в части ДНК, называемой SMN1 ген, который обычно производит белок SMN. Из-за мутации гена, у людей со SMA производится меньшее количество SMN белка, что приводит к потере моторных нейронов.
Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием. Тип 1 SMA проявляется у детей в период от рождения до 6 месяцев.
Пациенты способны сидеть без поддержки или даже стоять с поддержкой и обычно не страдают при приёме пищи. Однако они имеют увеличенный риск осложнений от инфекций дыхательных путей. Тип 2 SMA проявляется у детей в период между семью и 18 месяцами.
Пациент способен стоять, но испытывает сильную слабость, с тенденцией инвалидизации (передвижение в коляске). Тип 3 SMA проявляется после 18 месяцев, реже — во взрослом состоянии.
Признаки атрофии обычно начинаются в мышцах рук, ног и языка, и распространяются к другим областям тела.
Статистика приведена из Patient Registry Report, составитель Connie Garland для SMA Registry, The International Coordinating Committee for clinical trials in SMA:
Т.к. спинальная мышечная атрофия — нарушение, которое проявляется в синапсах моторных нейронов, состояние может быть улучшено за счёт увеличения уровня SMN - белка. Цель современных исследований - поиск препаратов, увеличивающих уровни SMN. Основные результаты получены пока в исследовательских группах США, Германии, Италии.
которые хотят иметь ребёнка или уже имеют ребёнка, диагностированного со СМА? Найдите медицинский центр, имеющий возможнось проведения анализа для установление предполагаемого носительства дефектного гена. Необходима пренатальная диагностика на ранних сроках беременности (до 14 недель). Помните, что на Вас лежит ответственность за здоровье будущего ребёнка.
В настоящий момент возможна только пассивная профилактика — предупреждение родителей с риском СМА о возможных последствиях, пренатальный скрининг — диагностика формы СМА для принятия решения о рождении. Диагностика производится в некоторых медико-генетических центрах.
в том числе распространенные заболевания, проявления которых которые могут быть скорректированы путем приема комплекса специальных лекарственных препаратов, диетой, БАД
Wikimedia Foundation. 2010.
dic.academic.ru














